|
2022/4
No.156 |
1. 巻頭言 | 2. コロナ照射法を用いたオキシ水酸アパタイトの電荷蓄積 | 3. ISO/IEC 17025 認定校正について | |||
|
|
||||||
<研究紹介>
![]() コロナ照射法を用いたオキシ水酸アパタイトの電荷蓄積
コロナ照射法を用いたオキシ水酸アパタイトの電荷蓄積
圧電物性デバイス研究室 安 野 功 修
1.まえがき
無機材料のエレクトレットは、1957 年深田等がストレスが骨に電位を発生させるという事実を圧電性に関連付けた[1]のがはじめで、その後、骨の主成分である Hydroxyapatite(HA)は生物医学的エレクトレットと
して生体適合性を備えているため、医療技術の革新に対する最も有望なアプローチの1つとして研究された。このHAは絶縁体ではなく、イオン伝導体であり、水酸アパタイト(Ca10(PO4)6(OH)2)のイオン伝導体中のキャリ アの動きを移動途中で凍結させてエレクトレットとしての性能を確保したものである。HAを使用して巨大な電 荷蓄積を達成する条件は、電界強度と分極に使用される
温度の両方に依存しており5.0 kVcm-1, 600 ℃の条件で 1.2 mCcm-2にまで達した[2,3]。本研究ではこの水酸化物イオン欠陥のあるHA:Oxy-hydroxyapatite(OHA)をエネルギー発電に利用するために、コロナ照射方式でOHA エレクトレットを作製し電荷密度、電荷保存安定度、熱刺激脱分極電流(Thermally stimulated depolarization current ;
以下TSDC と略記)測定による電荷トラップ 状態を従来の熱エレクトレット方式と比較、分極状態の一様性を評価した。
2 . 実験
2-1 OHA サンプル作成とコロナ照射処理
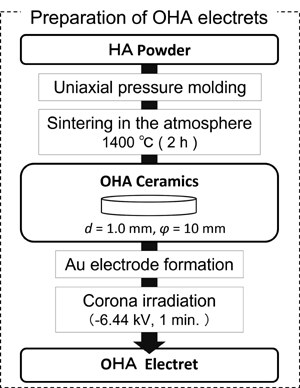
OHA セラミックサンプル作成のフローチャートを Fig. 1aに示す。HA粉末を型に入れ、一軸加圧成形によ りグリーンコンパクトを作製した。乾燥空気雰囲気下で、1400℃で2時間焼結した後、材料を室温まで冷却、ディスクの上面と下面を研磨し厚さ1
mmとした。さらにエタノールと純水で超音波洗浄し、両面に金電極を作製後、室温および通常の湿度(20 ~ 25℃、50 ~ 60%)のクリーンルームでコロナ照射により帯電させた。コロナ帯電条件の主なパラメータは、印加電圧と照射距離、照射時間、照射中の温度によって決定される。本研究で使用した電界と照射時間は、予備実験の結果に基づいて
決定した。電界により表面電位が上昇する傾向があるので、印加電圧を実験装置の最大値である- 6.4 kV に固定し、照射距離を2.0 cm(約3.2
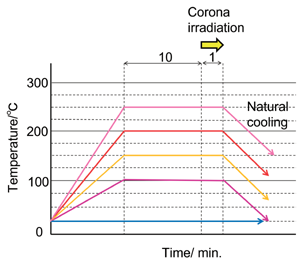
kV/cm)とした。照射時間は1分に固定、OHA 表面電位を飽和させるのに十分な値とした。ホットプレートを使用して、照射中の加 熱温度を25℃から250℃の間で制御した。詳細な温度プロファイルをFig.1b
に示す。コロナ照射中、異なる温度条件下で5種類のサンプルを準備した。
| a) |
 |
| b) |
 |
|
Figure 1a. Preparation of OHA.
Figure 1b. Temperature profile of corona irradiation. |
2-2 エレクトレットの特性評価
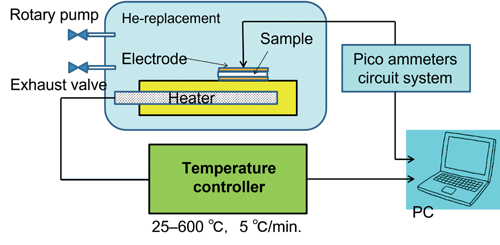
コロナ照射されたOHA セラミックの表面電位と電荷蓄積は、それぞれKelvin プローブとTSDCで評価した。TSDC 測定のセットアップをFig.2
に示す。サンプルを He ガスで満たされた真空チャンバーに入れ、TSDC は、25~600℃の範囲で温度制御し、加熱速度 5℃/分の短絡モードで測定した。
 |
|
Figure 2. Setup for automatic
thermally stimulated depolarization current measurements.
|
3.結果と考察
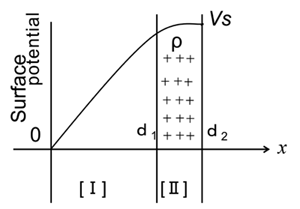
コロナ照射によるOHAセラミックの帯電挙動を予測するために、Fig.3に示すエレクトレットモデルにおいて電荷がHA 表面近くのx
= d1 とx = d2 の間で均一な密度で存在すると仮定して、電荷密度ρを制御するための理論検証をした。領域[I]および[II]の電位をφ1
, φ2 とした場合、V S は表面電位、εはHA の誘電率である。
 |
|
Figure 3. Schematic model of
charge storage and surface potential assumed for corona-irradiated
OHA ceramics.
|
  |
境界値条件は
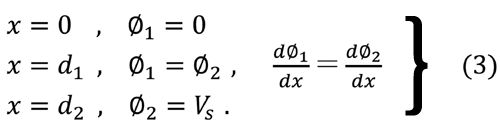 |
解は次のようになる。
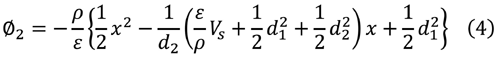 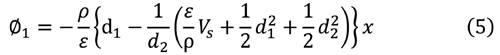 |
一方、ρの負電荷は x = 0 の表面に分布することになる。単位面積あたりの電荷Q は
 |
ガウスの定理より
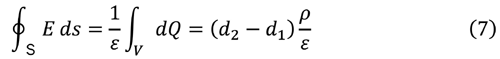 |
故に電界E は
 |
式 (5)と(8)より電荷密度 ρ は表面電位V S より
 |
たとえば、d1 = 0.5d2 の場合、V
S = 1 kV とすると荷 電後のOHA 容量は6.5 pF であるため、式(9) から電荷密度ρは 34.5μc
/ cm2 になる。ただし、d1 = 0.99d2
の場合、電荷密度ρは1.20 mc / cm2 と推定され、コロナ照射条件に応じてd1の値を変えることができれば、電荷密度ρを制御できる可能性がある。
OHA の結晶相は、X 線回折法(XRD)とフーリエ変換赤外分光法(FTIR)及び走査型電子顕微鏡(SEM)を用いて表面の形態を観察した。XRD
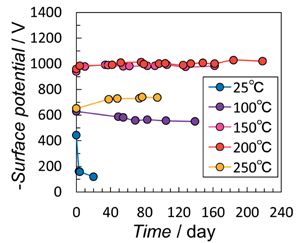
パターンと赤外線(IR)スペクトルに基づいて、化学量論的HAよりも 15%OH イオンを含む単相OHA として識別された。また、SEM 顕微鏡写真でディスクは著しく密で、サイズが約11μmの粒子を含むことを確認した。コロナ照射サンプルの表面電位測定は、表面電位の初期値と保持挙動
がコロナ照射中の加熱温度によって明らかに影響を受けることを示した(Fig.4)。150℃および200℃で処理されたサンプルは、25℃、100℃、および250℃で処理され
たサンプルよりも高い初期電位を示した。これは、照射直後に表面電位を上げるための最適な加熱温度の存在を 示唆している。250℃での表面電位の低下は、コロナ放電電荷の表面伝導の増加によるOHA表面の電荷密度の低下が原因であると考えられる。さらに、室温までの冷却時間を長くすることによる内部双極子の緩和も、表面電位の低下に寄与する。ただし、保持は加熱温度とともに増加した。25℃で処理されたサンプルの表面電位は
半分未満に急速に減少し、100℃で処理されたサンプルは数日以内に数パーセントの漸減を示したが、150℃を超えて処理されたサンプルは約220日間十分な保持を示した。Fig.
4 に示す各条件のデータは、同じ製造仕様でのいくつかの実験に基づいて再現性が確認された代表的なデータである。
 |
|
Figure 4. Temporal change in
surface potential of OHA ceramics corona-irradiated at varied temperatures.
|
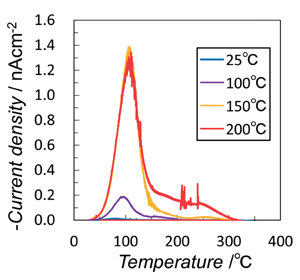
TSDCのピークは、すべてのサンプルで25~350℃で観察され、加熱温度の上昇とともに増加する傾向があった(Fig. 5)。TSDC 曲線から得られたQ 値は、式(1) から(9) で予測された値の20分の1であり、プローブが短絡したときに漏れが発生したか、電流が他の経路を流れたと推測される。Shunbo 等は密度汎関数理論計算の結果から、化学量論的HA の単斜晶(P21 / b)相と六角形(P63 / m)相の両方で強誘電分極を明らかにした[4]。また、Tofail等は機械的負荷と極性の間の方向性のあるリンケージの観点から、HAセラミックの巨視的スケールの圧電性を証明し、極性反転が300 ~ 500℃で発生する分極HA セラミックの焦電性を報告している[5-6]。コロナ照射されたOHAセラミックの場合、TSDC測定は 5℃ / 分で温度を25℃から600℃まで上げてHeガス中で自然冷却を行い(加熱速度よりも速い温度勾配で)測定した。しかし、冷却プロセス中に、TSDC 曲線は徐々にゼロに減少し、極性の反転は観察されなかった。
 |
|
Figure 5. TSDC curves of OHA ceramics corona-irradiated
at various temperatures (25-200 ℃).
|
温度T での双極子緩和電流は、次の式を使用して計算されることが知られている。
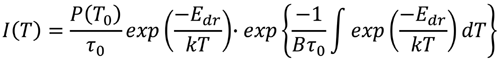 (10) (10) |
ここで、I(T)は温度T での双極子緩和電流で、P(T0)
は、温度T0での分極量、k はボルツマン定数、βは加熱速度、τ0は双極子緩和時間の前指数因子、Edrは双極子緩和の活性化エネルギーである。
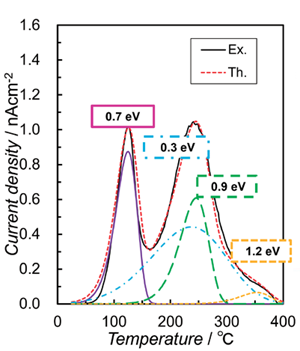
Fig. 6a は、熱エレクトレット法(VS ≅ 1 kV)で作成されたOHAエレクトレットに対して実施されたTSDC
分析の結果を示している。ただし、冷却プロセス中に電 流が検出されなかったため、加熱プロセスの結果のみを示した。実線と破線は実験値と計算値を表しており、
OHA エレクトレットには異なる活性化エネルギーEdr を持つ4種類のポーリングモードが存在することを示している。Shunbo およびTofail
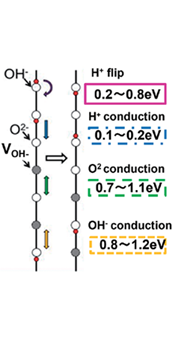
等の論文より[4-6]、HA の 強誘電性は、H+の局所的な回転によるc 軸内のOH-秩 序化によってもたらされると考えられている。一方、笠松等はFig.
6bに概略的に示すように、H+の局所的な反転だけでなく、プロトン、酸素、および水酸化物イオンの移動も、大きなOH-欠陥を持つOHAで比較的容易に発生する可能性があると述べている[7]。c
軸に沿って直線的に配置されたOH-サイトでの強誘電挙動(局所プロトンフリップ)に加えて、イオン移動によるものと考えられる。TSDC
分析で得られたEdr の値が第一原理計算で推定された活性化エネルギーとよく一致していることを考慮すると、OHA
分極の形成は、H+、O2- 、およびOH-イオンの移動によって実現されることが認識された。
H+の反転と同様に主にOHAエレクトレットの表面電荷に寄与すると思われる、移動によって引き起こされるイオン変位によって形成されるこのような「不安定な」双極子は、不均一な分布から均一な分布へのイオンの自己拡散により、TSDC
の加熱プロセス中におそらく完全に緩和される。ただし、冷却プロセス中でも、 OH-イオンの局所的な秩序がわずかに残っている場合はある。
|
a)
|
b)
|
 |
 |
|
Figure 6a. Experimentally obtained
TSDC curve (Ex.) of OHA electret
s prepared via thermal electret method with the theoretical curves (Th.) calculated using Edr values of 0.3 1.2 eV. Figure 6b. Schematic illustration of the four stage process estimated by first-principles calculation[7]. |
|
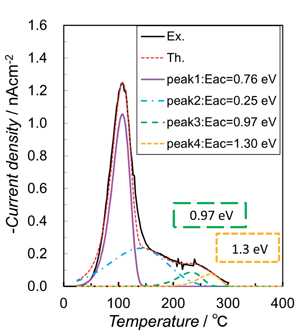
コロナ照射OHAセラミックの双極子形成と緩和メカニズムを詳細に理解するために、実験的に得られたTSDC 曲線を、複数の緩和プロセスを想定して式(10) を使用して理論ピークに適合させた。すべてのサンプルについて、200℃(V S ≅ 1 kV)で準備されたサンプルの結果を Fig. 7 に示す。4種類の熱緩和プロセスを想定して曲線を理論的に再現した。4段階プロセスでモデル化された熱緩和挙動は、熱エレクトレット法で得られた OHA エレクトレットの挙動と類似しており、荷電方法の違いに関係なく、Edr の値は各プロセスでほぼ同じであった。
 |
|
Figure 7. Experimentally obtained TSDC curve (Ex.) of OHA electrets prepared via corona irradiated
at 200 ℃ with the theoretical curves (Th.) calculated using Edr values of 0.25-1.3 eV.
|
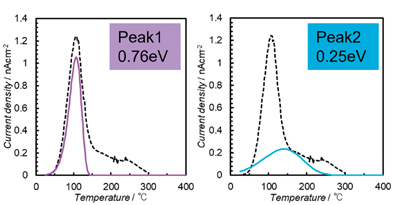
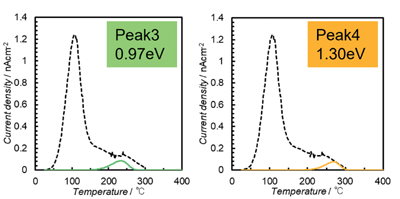
Fig. 8は、4つの活性化エネルギーでの緩和電流を個別に示した。4段階プロセスの最初の段階の100℃付近での緩和電流の大きな放出は、H+の反転によるものと推定され、OH-の局所秩序から解放された後、H+ とO2-および OH-は移動を開始する可能性があり、その結果、約300℃までの加熱中に完全な脱分極が生じる。240℃(ピーク3)および270℃(ピーク4)でのピークの低さは、コロナ照射中のO2-およびOH-の移動の不十分な活性化に起因する。このプロセス中、分極とTSDC測定を繰り返したOHAセラミックでTSDCの同じ曲線が得られ、外部への水の放出などの組成変化は発生していない。
  |
|
Figure 8. Four-stage process for OHA ceramics corona-irradiated at 200 ℃
|
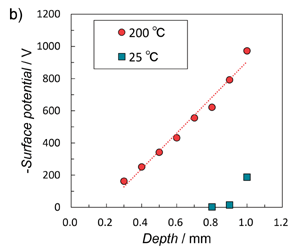
次に材料の深さ方向の帯電状態を評価した。Fig. 9a は、材料を深さ方向に0.1 mm カットした断面を示す。 Fig. 9b はそれぞれの表面電位の測定値を示している。表面電位は深さ方向に直線的に変化することがわかった。観測された電荷分布は、Fig.
3 に示したモデルの d1 ≅ d2
の条件に近い分布状態の電荷分布に似ていた。
 |
 |
|
Figure 9a. Cross section of
OHA
Figure 9b. Surface potential vs. depth within OHA sample. |
4.まとめ
100℃を超える温度でOHA セラミックをコロナ照射すると、イオン移動(H+、O2-、およびOH-)の活性化により、OHAセラミックに内部分極が形成された。分極は、熱エレクトレット法による観察結果と同じ4段階の分子移動プロセスによるものである。さらに、TSDC のピークは、コロナ照射の電界と温度条件に依存することがわかった。
コロナ照射エレクトレットは、処理時間の短縮、表面電位の制御など、OHA のエレクトレット化における問題を解決する方法として有用であることを確認した。
参考文献
[1] E. Fukada et al J. Phys. Soc. Japan, vol. 12, pp. 1158-1162, 1957.
[2] Y. Tanaka et al.,“Polarization and microstructural effects of ceramic
hydroxyapatite electrets”, J. Appl. Phy., vol. 107, p. 014107, 2010.
[3] S. Nakamura et al,“Proton Transport Polarization and Depolarization
of Hydroxyapatite ceramics,” J. Appl. Phys., vol. 89, no. 10, p. 5386 92,
2001.
[4] Shunbo Hu et al, “Ferroelectric polarization of hydroxyapatite from
density functional theory,” RSC Adv., 21375-21379, 2017.
[5] S. A. M. Tofail et al,“ Direct and ultrasonic measurements of macroscopic
piezoelectricity in sintered hydroxyapatite”, J. Applied Phys., 105, 064103,
2009.
[6] Tofail et al., “Pyroelectric surface charge in hydroxyapatite ceramics”,
J. Applied Phys.,106, 106104, 2009.
[7] S. Kasamatsu et al,“ First principles investigation of polarization
and ion conduction mechanisms in hydroxyapatite,” Phys. Chem. Chem. Phys.,
vol. 20, pp. 8744-8752, 2016.